This is a brief description of our software. You will find the following information:
- Whole Genome Alignment Problem
- Description of Our Software
- System Requirements and Estimated Running Time
- Sample Output of Our Software
Whole Genome Alignment Problem
With the completion of more and more genome sequencing projects, there is an increasing call for software tools that compare (align) two genomes. Given the genomes (DNA) of two related species, the whole genome alignment problem is to identify potential regions that contain genes and/or other regulatory elements that are conserved over the two species.
TopGiven two genomic sequences of related species (e.g., a pair of human and mouse chromosomes), our software reports the regions that are likely to be conserved over the two species.
We employ an approach similar to the one used in MUMmer -- a popular whole genome alignment software tool. The whole process is divided into three steps:
| Step 1. |
Finding matched substring pairs. |
| A pair of conserved regions over two species often contain many matched substrings that are unique to this pair of regions. In this step, we construct a program based on suffix tree to find these maximal uniquely matched substring pairs with length at least l, for some l > 0. Different values of l can be chosen for DNA and protein sequences. | |
| Step 2. |
A simple filtering. |
| As Step 1 often reports a large number of isolated matched substring pairs that are likely to be noisy pairs, we apply a filtering to remove some trivial isolated pairs. For each matched substring pair m, we check for the existence of another pair within a pre-defined distance d on the genomes. If no such a pair exists, we remove m. To avoid removing correct matched substring pairs, d should be set to a reasonable large value. | |
|
Step 3.
|
Clustering the matched pairs. |
| After filtering, we apply the MaxMinCluster algorithm (See References for details) to find an alignment of the remaining matched substring pairs. |
System Requirements and Run Timing
Unix/LinuxMemory Requirement:
- Identify MSPs
We use suffix tree to identify MSPs. This requires quite a large memory space, especially for long sequences. The following table gives a rough summary of memory.
(insert a table here)
- Cluster MSPs
The memory space needed for the clustering algorithms depends on both the number of input MSPs and the noise level. The following table gives a rough summary of memory.
(insert a table here)
Running Time: (based on a computer with 2.0GHz CPU).
- Identify MSPs
The time needed to identify the MSPs depends on the lengths of the two input sequences. In our testing, it takes around 1 hour to find MSPs between two sequences of 60M.
Length 1 (base pairs) Length 2 (base pairs) Time Taken (seconds) 1,000 1,000 0.153 10,000 10,000 0.271 100,000 100,000 9.607 - Cluster MSPs
The time needed for the clustering algorithms depends on to both the number of MSPs and the noise level. The following table gives a rough summary of the running time for the clustering algorithms.
# of Input MSPs Noise level (k) Running Time 30,000 0 0.5 hours 30,000 3 2 hours 70,000 3 8 hours 150,000 3 32 hours
Before Step 2
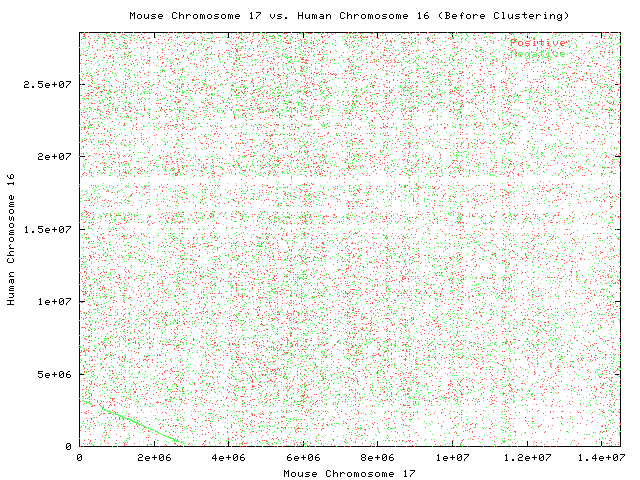
Each point represents a pair of maximal matched substrings
After Step 3
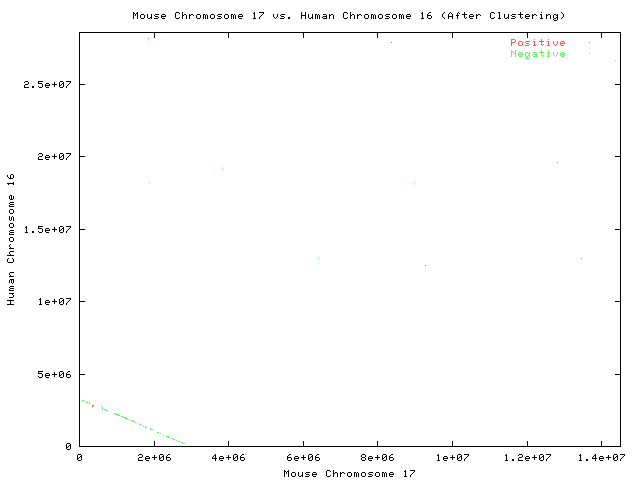
Each point represents a pair of maximal substrings.
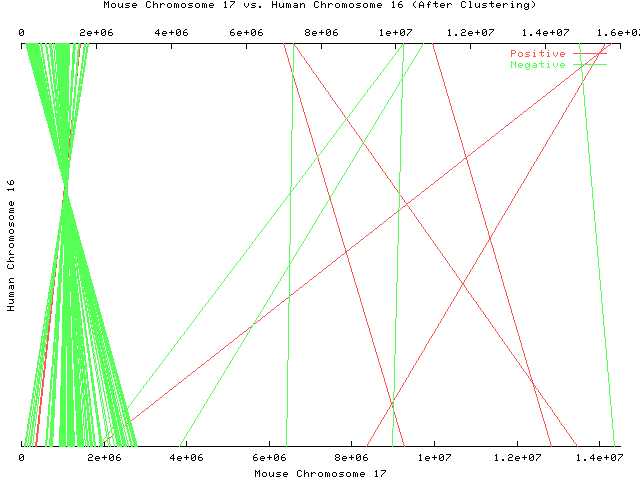
Each line represents a pair of maximal substrings.
Zoom in on a subregion of the two sequences (first 3,000,000 nucleotides of both sequences)
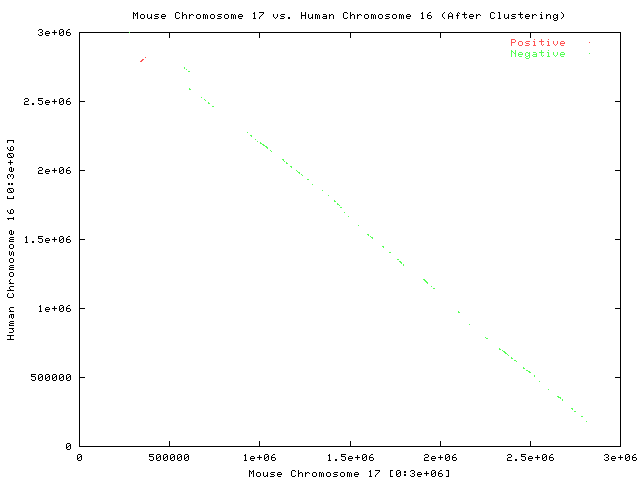
Each point represents a pair of maximal substrings
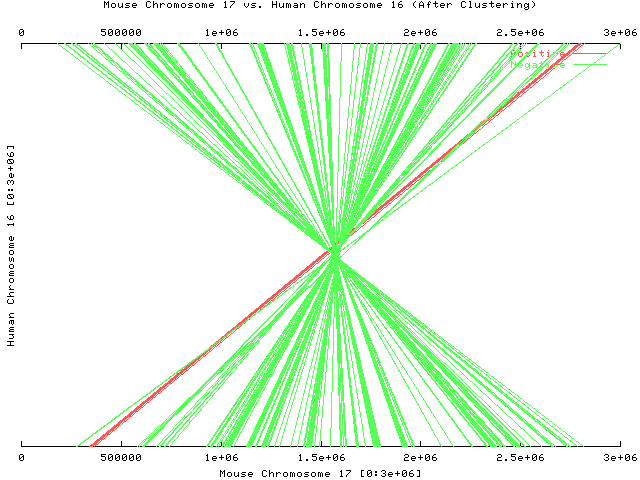
Each line represents a pair of maximal substrings